Introduction to the IVDR
Introduction to the IVDR

Fay Nijenhuis
Junior Consultant at NAALA
Published on 10 June, 2022
By: Fay Nijenhuis – Junior Consultant at NAALA
Published on 10 June, 2022
May 26th, 2022 was the date that the In Vitro Diagnostic Medical Devices Regulations (hereinafter: “IVDR”) became applicable. Before this date, the European Directive 98/79 EC (IVDD) was the valid directive from 1998 to 2022. The IVDR therefore replaces the IVDD, with new and enhanced rules. What changes have been made regarding these two kinds of legislation?
What is the definition of an In Vitro Diagnostic Medical Device?
In vitro diagnostic medical devices (IVDs) contain all equipment, software and reagents that are intended to investigate human specimens. Human specimens are, for example: blood, urine, cerebrospinal fluid, tissue and cells. A few examples of IVDs are: Containers for collecting and keeping bodily samples, blood glucose meters and nowadays the very well-known Covid-19 self-tests. The purpose of the information can be found in one or more of the following:
Concerning a physiological or pathological process or state. For example, body temperature, polycystic ovarian syndrome (PCOS) or sleep-wake cycles.
- Concerning congenital physical or mental impairments i.e., birth defects, such as a hole in the heart, a valve problem, a problem with blood vessels, as well as emotional illnesses or bipolar disorders.
- Concerning the predisposition to a medical condition or a disease i.e., the likeliness to have a specific illness.
- To determine the safety and compatibility with potential recipients. For example, to ensure the blood type of the donor matches the blood type of the recipient.
- To predict treatment response or reactions. For example, companion diagnostics to identify patients likely to be at increased risk for serious side effects as a result of treatment.
- To define or monitor therapeutic measures i.e., patients who need specific drugs to relieve pain or treat infections.
The world of medical devices is of great importance, with more than half a million medical devices on the European market. In comparison to the 10.000 chemical entities in medicines, most people underestimate the extent of this market. Furthermore, technological developments for innovative medical devices are skyrocketing. To ensure the quality of each (new) in vitro medical device, the European Union implements the IVDR and increases patient safety.
The need for transparent regulations is caused by, unfortunately, some scandals in the world of medical devices (read our blog post: “Rolling Barrels and Ruptured Breast implants”). Therefore, the EU primarily revised the regulations regarding medical devices (MDR), as well as the IVDR recently. See our blog post for a clear overview of the differences between the EU MDR and the EU IVDR on applicability, pre-market data, post-market data and the role of the notified bodies.
If you are a manufacturer, authorized representative, importer or distributor of IVDs in the European Union, the IVDR is applicable for your business operations.
The transition from the IVDD to the IVDR brought several changes to the medical device world. We will describe three significant changes and compare these to the old legislation.
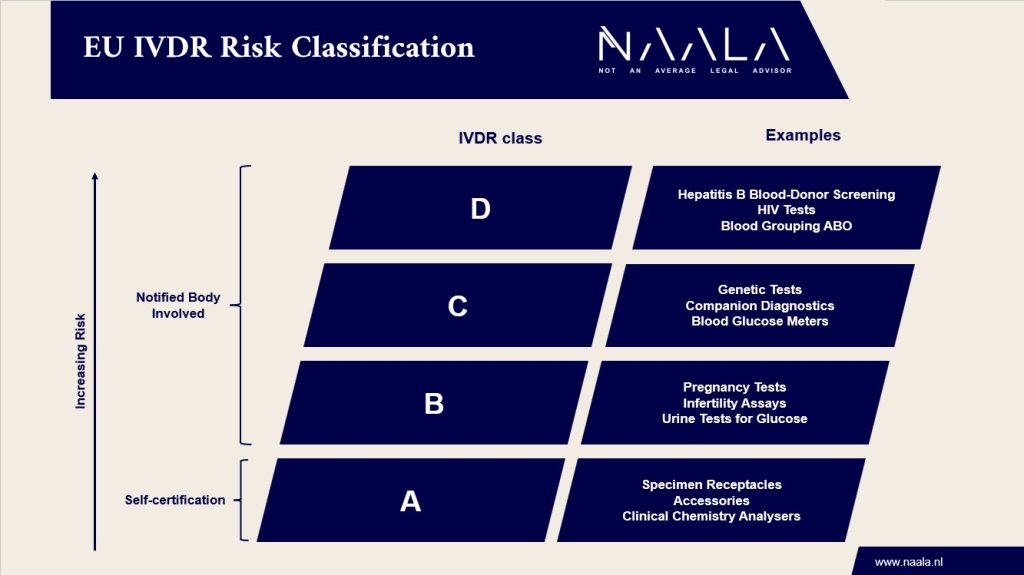
First, a new risk classification scheme was introduced. The IVDR has 4 risk categories ranging from A (lowest risk) to D (highest risk). The higher the risk classification, the stricter the requirements. IVDs from class B and higher must be certified by a Notified Body (an independent third party), whereas manufacturers of Class A IVDs still may perform self-certification. Under the old legislation, a so-called list-system was used. Lists were drawn up, indicating which types of IVDs were considered high risk. Approximately 80 percent of the IVDs were classified in the low-risk category, the self-certification category. With 4 risk categories under the IVDR, more IVDs will be in class B or higher and therefore have to involve a Notified Body. An estimated 85 percent of all IVDs will have to involve a Notified Body from now on, due to the new classification scheme. This may lead to a significant increase in workload in terms of certification processes, as well as a predicted increase in the quality of the IVDs.
Please see our visual overview at the bottom of this blogpost.
Second, manufacturers have to carry out more Post Market Surveillance (PMS) activities. PMS is a comprehensive process for monitoring and assessing the safety and performance of medical devices and IVDs. The process starts before the device is placed on the European market and continues for the entire life cycle of the device. This guarantees the safety of the products after being marketed, by contacting the users and patients on a regular basis. Besides the regularity, the PMS also needs to be active, i.e. not just waiting for complaints to be recorded. Lastly, the PMS needs to be documented i.e., an exact description of processes and results. These three points are important because Notified Bodies are required to carry out audits at least every 12 months – including an assessment of PMS planning, data analysis and reporting. By carrying out more PMS activities, the quality of the product throughout its entire lifetime will be guaranteed.
Third, a European database with information about IVDs on the European market has been generated. This database is better known as EUDAMED. EUDAMED is used to inform patients and healthcare professionals about the IVDs so that informed choice can be made for treatments. Post Market Surveillance data has to be transferred to EUDAMED to ensure the safety of the IVD. The side effect is that processes will cost more time and money since the applicability of the IVDR. For manufacturers it is therefore important that personnel is very well trained and the Notified Body has to be informed as quickly as possible if any significant change appears, which may result in additional burden.
Regulations are part of the European legislation with a direct effect. In contrast to directives, regulations do not have to be converted into national legislation. That means that citizens and companies can directly derive rights from the IVDR but are also obligated to comply with the rules. The IVDD was a directive and needed to be converted into national legislation. The objectives of a directive must be reached by all EU countries and translated into their national legislation within a defined time frame. However, it is up to all individual countries to define how to achieve the objectives by implementing them in their national laws. The consequence: the directive may be enforced in different manners in the several EU Member States and these differences can cause regulatory difficulties.
With the entry of the IVDR, the rules have been given the form of a regulation instead of a directive. A regulation is a binding legislative act and is immediately applicable in all EU Member States. It overrules the national laws and results in every EU country applying the exact same rules. For example, an IVD made in Italy or reviewed by an Italian Notified Body complies with the same quality objectives as an IVD made in the Netherlands or reviewed by a Dutch Notified Body.
Questions? We are happy to discuss your specific case.
Related
Do you manufacture facial fillers? You might be developing a medical device. The industry of facial or other dermal fillers have faced several regulatory changes over the year. Historically, dermal fillers have been marked as medical devices, medicinal product, or cosmetics. With the applicability of the EU MDR last May, the EU has tightened the requirements applicable to facial fillers. Following the EU MDR, facial fillers qualify as medical devices.
The arrival of the MDR brings changes to the entire medical device market: from raw material suppliers to healthcare consumer or patient. However, the biggest responsibility (still) lies with the manufacturer. The manufacturer must demonstrate that the device meets its requirements. What exactly is the difference between the MDD and the MDR? What do you need to do as a manufacturer to ensure MDR compliance?
Please note that all details and listings do not claim to be complete, are without guarantee and are for information purposes only. Changes in legal or regulatory requirements may occur at short notice, which we cannot reflect on a daily basis.